为什么需要分子对接
当我们需要筛选一堆药物分子针对某个靶点(蛋白质)的结合效果哪些分子更好,或者我们在判断多个蛋白质突变体分析和一个分子的结合效果哪些突变体更好时,我们就需要分子对接这种技术。
需要什么软件
- Anaconda
- Pymol
- Openbabel
- MGLTools
- Autodock Vina
- (对于设备而言,一般AutoDock Vina只占用CPU进行计算,但计算速度很快,本实例轻薄本11代核显i5即可轻松运行)
安装
安装pymol
打开Anaconda Powershell Prompt输入(将以下命令以此输入到命令行中)
conda create -n pymol
conda activate pymol
conda install -c conda-forge -c schrodinger pymol-bundle
安装openbabel
- 上一步完成之后输入
conda deactivate
或者直接退出命令行重新进入
- 输入
conda create -n openbabel
conda activate openbabel
conda install conda-forge::openbabel
安装MGLTools和Autodock Vina
进入MGLTools和Autodock Vina分别下载安装MGLTools和Autodock Vina(一定要安装在独立的文件夹中)。
分子对接基本流程——以c-Abl蛋白和格列卫(imatinib)为例
所需材料
- 受体(蛋白质)的坐标文件
- 配体(小分子)的坐标文件
受体及预处理
在PDB (Protein Data Bank)中下载本次实例所用到的1iep.pdb文件。请提前将MGLTools中的adt.bat和AutoDock Vina中的vina.exe复制到了受体配体同一文件夹中。此时对接文件夹中文件如图: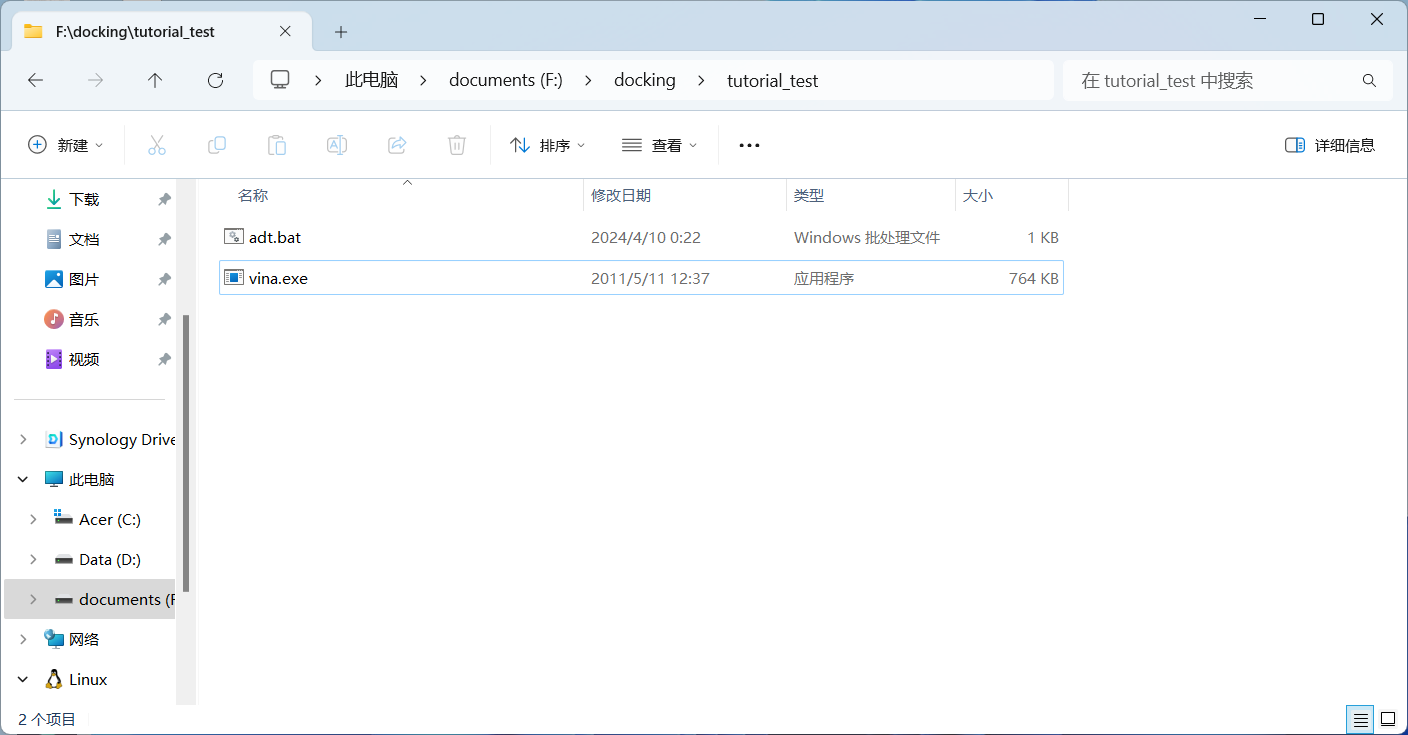
- 去除杂原子:这一步需要使用pymol,并且查阅文献确定删去哪些杂原子。如果该蛋白需要这些辅酶或离子才能发挥作用的话,那就不可以删去,以保证对接的准确性。有些分子/离子的存在是由于为了方便蛋白质结晶而引入的,此时这些杂原子就应该删去。以
1iep.pdb为例,在pymol中观察可以发现该晶体为蛋白-配体复合物,且该配体实际上是和我们准备的配体一致的,很容易判断其结合的位点就是后续我们要结合的位点,如果不去除,会直接影响到对接。此外,通过pymol可以观察到该文件中还有6个氯离子。在Google Scholar中搜索 : “c-Abl” AND (“Cl” OR “chloridion”)。结果中没有提到氯离子,所以初步判断氯离子也是为了便于结晶而引入的,可以删去(对于不确定的杂原子,可以使用不同的情况多次对接比较结果)。在pymol中执行Display->sequence即可看到整个文件中包含的原子,选中需要删除的杂原子,点击右侧(sele)栏的A选项->点击”remove atoms“即可。 - 除水:Edit→delete water; 由于蛋白质晶体结构文件一般来源于X射线衍射晶体蛋白质,收集衍射图谱后计算蛋白质的原子结构,所以不可避免地会掺入一部分水分子。为了分子对接的准确性,我们需要使用MGLTools中的AutoDockTools(ADT)进行除水(这一步也可以使用pymol)。
- 加氢:Edit>Hydrogens>Add>OK; 在X射线衍射中,很难观察到氢原子的信号,所以获取的pdb文件不含氢原子。而加氢是为了描述受体蛋白的化学性质。比如说很多酶催化的能力来源于活性位点中某些极性残基的活泼氢(如天冬氨酸的羧基氢)或氢键作用(如苏氨酸的羟基氢)。所以我们要使用ADT给受体加氢,尤其是极性氢,这也就是很多教程中写只加极性氢的原因。不过为了结果准确,非常建议加入全部的氢原子。
- 计算电荷:Edit->Charges; 受体和配体的电荷在很大程度上影响着二者的相互作用,自然严重影响着对接结果,所以需要计算受体的电荷。在ADT的Edit->Charges中可以看到有两种计算电荷的方法,Compute Gasteiger和Add Kollman Charges。对于一般分子对接而言,Compute Gasteiger的精度就已经足够,其优点在于速度快。而Kollman方法适用于对于精度要求很高的作业,缺点在于所需时间太长。当使用Gasteiger之后结果不符合预期,再考虑使用Kollman。
- 添加原子类型:Edit->Atoms->Assign AD4 type; 除了以上三种预处理操作之外,还需要知道各个原子的类型,以及之间的相互作用。在添加原子类型的时候,也在为原子添加相应的力场参数(键长,键角等)。
- 保存为.pdbqt文件:Grid->Macromolecule->Choose->Select Molecule->确定->保存即可,此时文件夹中就多了一个
1iep.pdbqt文件。
配体及预处理
实例中,配体为imatinib,这是一种成品药物,在PubChem中搜索imatinib,下载3D Conformer的SDF文件(也可以使用ChemDraw绘制所需分子的结构并且保存为mol2文件)。
- 文件转化:使用Openbabel。进入openbabel的conda环境中,使用命令
obabel 'path/to/your/input/sdf/file' -O 'path/to/your/output/pdb/file'
- 添加配体:Ligand->Input->Open; 该操作后,ADT会自动完成计算电荷-分配原子类型的操作,所以要确认在Open之前,该配体氢原子的添加无误。
- 判定配体的root:Ligand->Torsion Tree->Detect Root; 识别配体分子中旋转结点,方便之后在对接过程中,分子依据此进行旋转,找到最佳的结合构象。
- 选择配体可扭转的键:Ligand->TorsionTree->Choose Torsions->Done; 在此步骤中,一般不需要做其他操作。除了某些特别的情况需要指定其中某些键可以旋转,某些键不可以旋转。
- 保存为.pdbqt文件:Ligand->Output->Save as PDBQT
对接准备
在进行这一步时ADT中的分子应该删除,为了保证思路清晰。
- 打开受体:Grid->Macromolecule->Open
- 打开配体:Gid->Set Map Types->Open Lignd
- 设置对接盒子:Grid->Grid Box; 手动调节滚轮将三色盒子包裹住受体的结合位点。为了确定结合位点,可能需要参考文献,参考包含配体的晶体结构,参考一些结合位点预测软件的结果。如果无法确定,也可以简单地将盒子包裹住整个蛋白。调整好之后,在Grid Options页面点击File->Close Saving Current
- 输出配置文件:Docking->Output->Vian config(config.txt); 在Vina Input Parameters页面中选择Show options,为了结果的准确,可以将modes. 和 exh. 的数字改大一些(在本实例中,
num_modes为50,exhaustiveness为200)。Save之后需要手动检查文件夹中config.txt中的参数是否符合要求,不符合要求需要手动加入。具体的参数设置请参考这里。本实例的配置文件如下: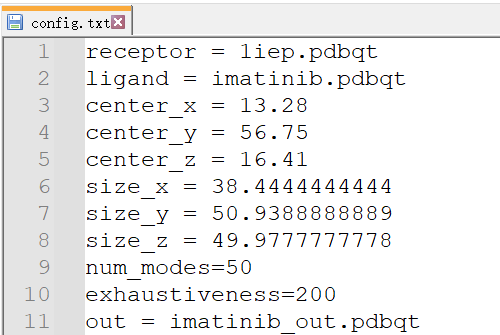
- Double Check:请保证将MGLTools中的
adt.bat和AutoDock Vina中的vina.exe复制到了受体配体同一文件夹中。此时文件夹中的文件如图: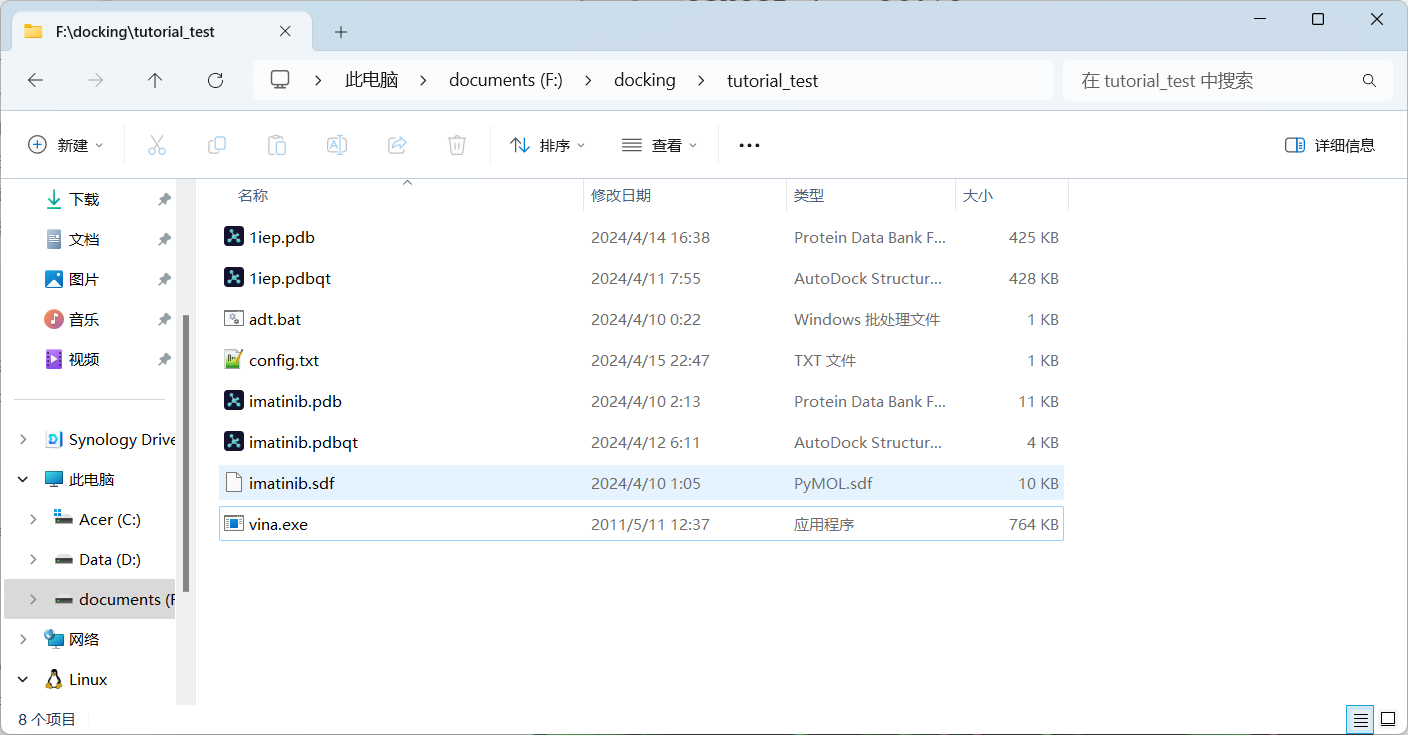
- 运行:可以通过命令行在工作目录下使用
./vina --config config.txt
运行,也可以在ADT内部通过Run->Run Autodock Vina->Launch来运行。
运行结束之后自动将num_modes个结果打印在屏幕上。
- 分析结果:在pymol中同时打开
imatinib_out.pdbqt和原始包含配体的1iep.pdb,可以查看所有结果中各个构象和晶体中配体构象的重合情况。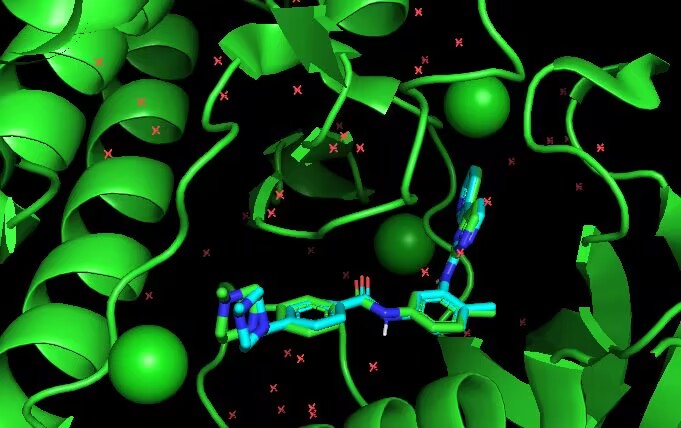
比对文献中分子对接的自由能和输出的结果,输出结果为-13.0kcal/mol为理想结果。
常见问题
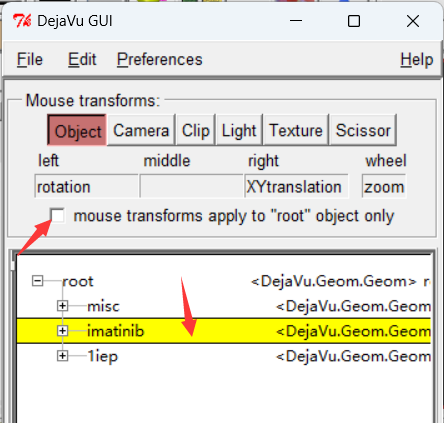
- 在对接准备阶段,打开受体配体文件之后发现二者重合。应打开ADT上方工具栏中的DejaVu GUI,选中小分子(或受体)一栏后取消选中mouse transforms apply to “root” object only选项。此时可以鼠标右键拖动小分子(或受体)。调整位置之后重新选中mouse transforms apply to “root” object only选项即可。
参考文献
转载请注明来源,欢迎对文章中的引用来源进行考证,欢迎指出任何有错误或不够清晰的表达。可以在下面评论区评论,也可以邮件至 1971945941@qq.com